
Twelve years ago, after a routine check-up of a newborn, a medical resident inadvertently ordered a nonroutine blood test. The results came as a surprise: The infant had virtually no neutrophils, a type of white blood cell that fights infections and heals wounds.
The physicians sought the cause of the child’s condition, called neutropenia. But as events unfolded, they spurred a remarkable odyssey steered by Raffaele Renella, then an attending staff physician at Boston Children’s Hospital, and David Williams, a renowned hematologist at Boston Children’s and the Dana-Farber Cancer Institute.
The duo unearthed a rare mutation in a gene that encodes a structural protein never before associated with neutropenia. Uncovered, the mutation provides doctors with a new understanding of the evolution of bone marrow failure and how some leukemias could develop.
Mireia Solá, formerly a postdoctoral researcher at the Institute for Protein Innovation (IPI) and a study co-author, used protein modeling software Alpha Fold 2 to clinch the link between gene and disease.
“What I really find exciting is we are not studying one disease, we’re not studying one patient,” said Renella. “But because of one patient and one disease, we opened a whole new chapter in biology.”
Leukemia, in real time?
When Renella, now a pediatric hematologist and oncologist at the University of Lausanne in Switzerland, met the infant in 2010, doctors did not yet suspect cancer.
They ordered tests to probe other illnesses that could have caused the neutropenia. But the results only yielded new puzzles: oversized red blood cells and high levels of fetal hemoglobin, possibly a sign that the newborn’s bone marrow — which should have been producing trillions of healthy blood cells each hour — may be failing.
The researchers looked at the bone marrow beneath a microscope, confirming their fears.
“It was unlike anything either Dr. Williams or myself has ever seen,” Renella said. “Something had gone terribly wrong.”
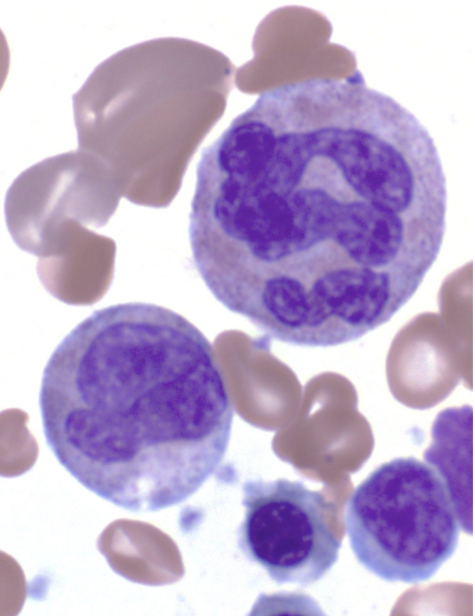
The infant’s neutrophils, not yet fully formed, were sparse in number and enormous in size, many packed with quadruple the normal amount of genetic material. Oddly, the stuffed cells were evading the usual cell-suicide programs meant to prevent defective cells from dealing damage.
Alarmingly, the bone marrow progressively showed telltale signs of pre-leukemia.
Physicians at Boston Children’s arranged a bone marrow transplant in the hopes of preventing leukemia. The transplant worked, but it also erased the culprits: What caused the cells to go rogue?
Gene-hunters in search of the cancer’s cause
Leukemia is the most common cancer in children and adolescents. It is not one disease but a group of cancers of the body’s blood-forming tissues, most often those that bear white blood cells, including the bone marrow and lymphatic tissue. Cells start out healthy but accumulate mutations over time.
Despite remarkable progress, exactly how those mutations mechanistically play out has long baffled cancer researchers. Part of the problem is that children diagnosed with leukemia do not show up in pediatricians’ clinics until they experience symptoms of advanced disease. That would have been the story for Renella’s patient if not for the early blood test.
“We thought, ‘Isn’t that just amazing? We have in front of us a patient who is showing us how leukemia may develop,’” Renella recalled.
The question was how to do the forensics. The transplant had corrected — and therefore erased — the mutated neutrophils.
Intent on an answer, Renella and the team began by conducting functional protein tests, hunting for mutations in the usually suspect genes. When they found nothing, they broadened the search to every gene previously linked to neutropenia. It was still a dead end.
Finally, they combed the patient’s entire genome — and found something: a mutation in the SEPT6 gene, located on the X chromosome. The gene codes for a structural protein called septin-6, which helps orchestrate the final steps of cell division. SEPT6 had already been linked to acute myeloid leukemia, especially in infants.
The infant’s SEPT6 gene had a stop-loss mutation — named for the loss of a stop codon, or a period that ends the sentence of a portion of genetic code — that lengthened the protein by nine amino acids.
In addition to that mutation, the researchers found something else troubling: In some cells, where the stop-loss mutation would have elongated the protein, a second stop-gain mutation cut the sentence off early.
Trying to find the origin of the mutation, the team sequenced the gene from the patient’s parents. Surprisingly, they didn’t harbor the stop-loss mutation. Instead of inheriting it, the infant likely developed it during a cell’s division in the first weeks of his intrauterine development. The stop-gain mutation came later, probably from a diseased white blood cell trying to recover from the initial mutation.
The findings suggest that, in some patients, early genetic mutations possibly linked to leukemia could be obscured over time by others.
“We do believe there are lots of situations like that, where patients have mutations that contribute to unhealthy bone marrow cells developing until the leukemia develops, and then become undetectable,” Renella said. “It is also possible that some of these mutations are effectively transmitted from the parents.”
Even with the mutations identified, however, the case wasn’t solved. How did the stop-loss mutation cause neutropenia? And how could that relate to bone marrow failure and pre-leukemia?
Too deadly for a lab demonstration
Studying the mutation would have usually required healthy blood cells from the infant’s bone marrow. Those cells, already scant, had been wiped out by chemotherapy before the transplant. Undaunted, scientists attempted to model the early stages of the patient’s condition in live cells.
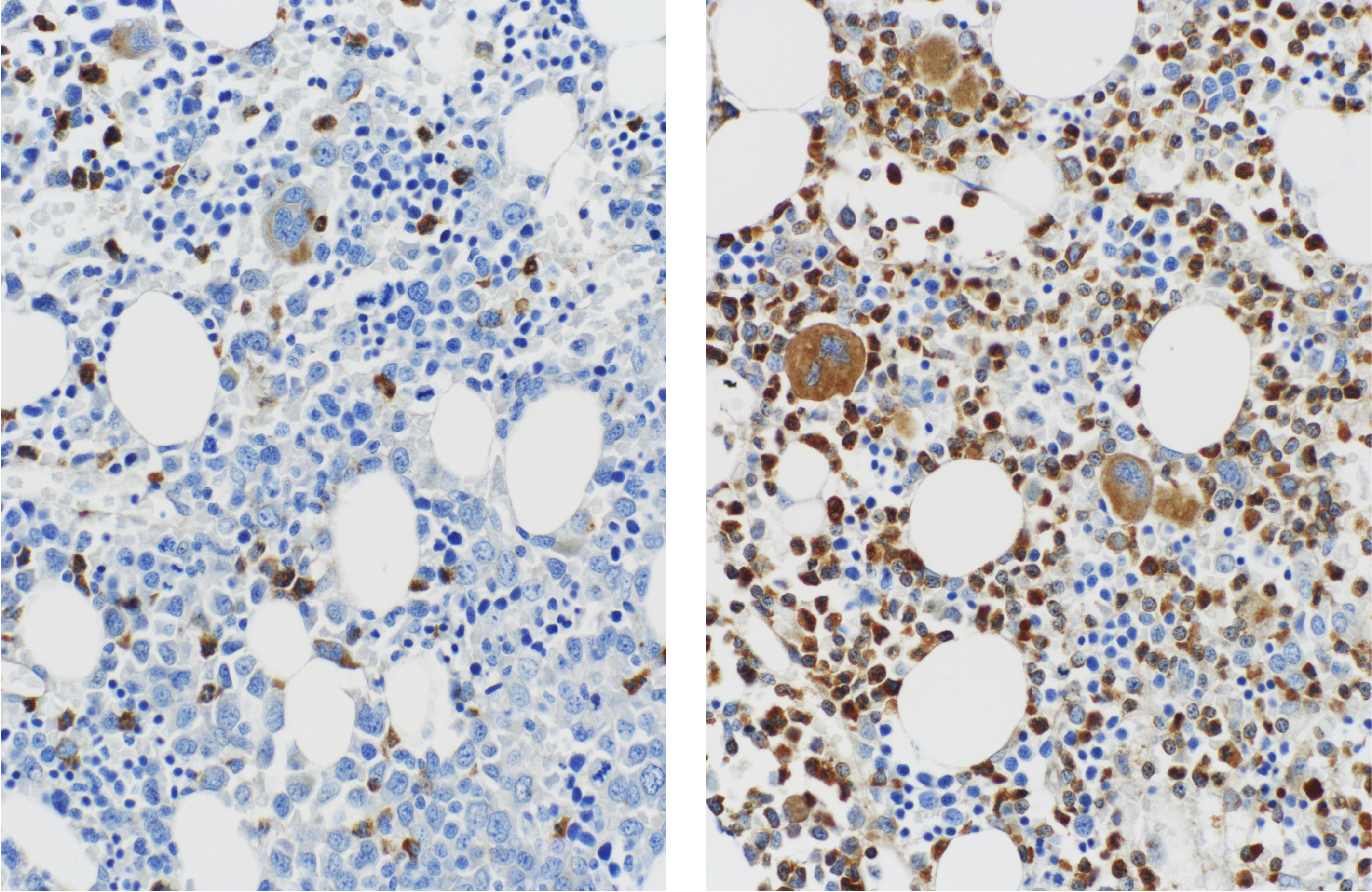
somatic tetraploidy due to a germline mutation in SEPT6” by Renella, R. et al.
They started with a then-novel technique, using self-renewing, induced pluripotent stem cells (iPSCs) that could be guided from skin cells into blood cells. Researchers found that the infant’s skin cells could be coaxed into other blood cells, but not neutrophils. By contrast, a healthy child’s skin cells could.
Next, the team turned to the gene-editing technique, CRISPR-Cas9, to insert the SEPT6 mutation into healthy cells. The defect again proved too deadly in white blood cells. They simply could not form neutrophils because the mutation was present.
“Every time you touched this protein in that vulnerable spot,” Renella said, “it would cause the cells linked to neutrophil production to die.”
Then, Renella heard about a study, this one on Shwachman-Diamond syndrome, another rare, heritable disease. Christopher Bahl, previously head of IPI’s Protein Design Laboratory, helped model the genetic mutation that led to the condition. Renella wondered if the same could be done for SEPT6.
He sought help from Bahl, who put Solá, a computational biologist, on the case. She deployed AlphaFold 2, a game-changing computational program taking the protein world by storm. She successfully modeled the likely shapes of the mutated septin-6, calculating the interactions among amino acids comprising it.
The model revealed that the nine extra amino acids researchers found on the C-terminus of septin-6 folded into a helix, creating a structured tail. That tail, Solá predicted, would hamper the assembly of single septins into supportive scaffolds, likely jamming cell division machinery. The chaos would leave the cells stuffed with copies of their DNA yet unable to divide.
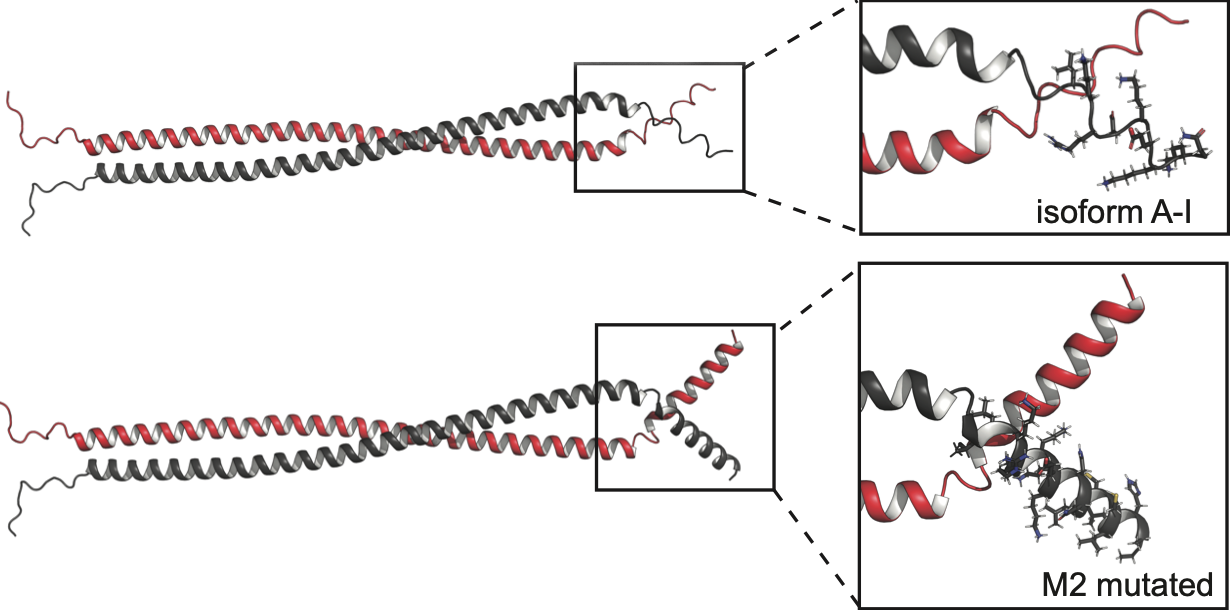
“This could explain the loss of function. And it had been our hypothesis,” Solá said.
It’s the first time septins have been linked to blood cell production, marking a new chapter in biology and, potentially, for early leukemia detection in patients with abnormal white blood cells.
“Every purely genetic investigation had been exploited, and every stone had been turned 15 times,” Renella said. “With Chris and Mireia and the IPI, we found a home for our doubts.”
Getting it right and early
Now, Williams, Renella and their team are working to identify new patients with similar mutations. Eventually, scientists could develop therapies for children predisposed to blood cancers because of bone marrow failure. Those therapies could enable mutated cells to divide or prevent them from propagating if they can’t, ultimately preventing leukemia.
The information is potentially life-saving. For other cases of undiagnosable neutropenia, doctors can include SEPT6 in their early screens and discuss bone marrow transplants before leukemia develops.
That knowledge came only through a decade of questions and the tireless pursuit of answers, the clincher coming from new technology proffered by impact-minded scientists at IPI.
“If you’re not curious, you’re going to look for the easy and simple,” he said. “But things are not easy. They’re not simple. That’s why nature is so beautiful.”
Writer: Halle Marchese, halle.marchese@proteininnovation.org
Sources: Raffaele Renella, raffaele.renella@chuv.ch;
David Williams, david.williams2@childrens.harvard.edu;
Mireia Solá, mireia.sola@aiproteins.bio